More cardiovascular disease occurs in patients with either type 1 or 2 diabetes. The link between diabetes and atherosclerosis is, however, not completely understood. Among the metabolic abnormalities that commonly accompany diabetes are disturbances in the production and clearance of plasma lipoproteins. Moreover, development of dyslipidemia may be a harbinger of future diabetes. A characteristic pattern, termed diabetic dyslipidemia, consists of low high density lipoprotein (HDL), increased triglycerides, and postprandial lipemia. This pattern is most frequently seen in type 2 diabetes and may be a treatable risk factor for subsequent cardiovascular disease. The pathophysiological alterations in diabetes that lead to this dyslipidemia will be reviewed in this article.
Causes of lipoprotein abnormalities in diabetes
Defects in insulin action and hyperglycemia could lead to changes in plasma lipoproteins in patients with diabetes. Alternatively, especially in the case of type 2 diabetes, the obesity/insulin-resistant metabolic disarray that is at the root of this form of diabetes could, itself, lead to lipid abnormalities exclusive of hyperglycemia.
Type 1 diabetes, previously termed insulin-dependent diabetes mellitus, provides a much clearer understanding of the relationship among diabetes, insulin deficiency, and lipid/lipoprotein metabolism. In poorly controlled type 1 diabetes and even ketoacidosis, hypertriglyceridemia and reduced HDL commonly occur .
Replacement of insulin in these patients may correct these abnormalities, and well controlled diabetics may have increased HDL and lower than average triglyceride levels.
The lipoprotein abnormalities commonly present in type 2 diabetes, previously termed noninsulin-dependent diabetes mellitus, include hypertriglyceridemia and reduced plasma HDL cholesterol. In addition, low density lipoprotein (LDL) are converted to smaller, perhaps more atherogenic, lipoproteins termed small dense LDL.
In contrast to type 1 diabetes, this phenotype is not usually fully corrected with glycemic control. Moreover, this dyslipidemia often is found in prediabetics, patients with insulin resistance but normal indexes of plasma glucose . Therefore, abnormalities in insulin action and not hyperglycemia per se are associated with this lipid abnormality. In support of this hypothesis, some thiazoladinediones improve insulin actions on peripheral tissues and lead to a greater improvement in lipid profiles than seen with other glucose-reducing agents .
Several factors are likely to be responsible for diabetic dyslipidemia: insulin effects on liver apoprotein production, regulation of lipoprotein lipase (LpL), actions of cholesteryl ester transfer protein (CETP), and peripheral actions of insulin on adipose and muscle.
Insulin regulation of liver apoproteins and lipid-metabolizing proteins
A number of studies using tracer kinetics in humans have demonstrated that liver production of apolipoprotein B (apoB), the major protein component of very low density lipoprotein (VLDL) and LDL, is increased in type 2 diabetes. ApoB is a large (>500-kDa) protein whose production is not modulated at the level of protein synthesis. In animals and cultured liver cells, transcription of the apoB gene is not remarkably altered by dietary changes and diabetes. Rather, a large amount of newly synthesized protein is degraded either during or immediately after translation. This degradation is prevented when lipid is added to the protein; this occurs via the actions of microsomal triglyceride transfer protein (the protein that is defective in patients with apobetalipoproteinemia). Thus, lipid regulates apoB production. Increased lipolysis in adipocytes due to poor insulinization results in increased fatty acid release from fat cells. The ensuing increase in fatty acid transport to the liver, which is a common abnormality seen in insulin-resistant diabetes, may cause an increase in VLDL secretion. Tissue culture , animal experiments , and human studies suggest that fatty acids modulate liver apoB secretion.
A second regulatory process may be a direct effect of insulin on liver production of apoB and other proteins involved in degradation of circulating lipoproteins. In some studies insulin directly increased degradation of newly synthesized apoB . Therefore, insulin deficiency or hepatic insulin resistance may increase the secretion of apoB. Insulin may modulate the production of a number of other proteins that affect circulating levels of lipoproteins. These include apoCIII (9), a small apoprotein that may increase VLDL by preventing the actions of LpL and inhibiting lipoprotein uptake via the LDL receptor-related protein (LRP).
Hepatic lipase is an enzyme synthesized by hepatocytes that hydrolyzes phospholipids and triglycerides on HDL and remnant lipoproteins. Some , but not all , studies suggest that this enzyme is reduced by insulin deficiency. One effect of hepatic lipase deficiency is to decrease the clearance of postprandial remnant lipoproteins (see below).
LpL is the major enzyme responsible for conversion of lipoprotein triglyceride into free fatty acids. This protein has an unusual intercellular transport; LpL is synthesized primarily by adipocytes and myocytes, but must be transferred to the luminal side of capillary endothelial cells, where it can interact with circulating triglyceride-rich lipoproteins such as VLDL and chylomicrons (13). Humans with both type 1 and type 2 diabetes have been reported to have reduced LpL activity measured in postheparin blood ; the enzyme is released from the capillary walls and into the circulation by heparin. Several steps in the production of biologically active LpL may be altered in diabetes, including its cellular production and possibly its transport to and association with endothelial cells.
LpL is stimulated by acute and chronic insulin therapy . LpL activity is low in patients with diabetes and is increased with insulin therapy .
The release of stored fatty acids from adipocytes requires conversion of stored triglyceride into fatty acids and monoglycerides that can be transferred across the plasma membrane of the cell. The primary enzyme that is responsible for this is hormone-sensitive lipase (HSSL). HSSL is inhibited by insulin, which decreases phosphorylation of HSSL and its association with the stored lipid droplet .
Specific lipoprotein abnormalities
Postprandial lipemia. Compared with normal subjects, patients with type 2 diabetes have a slower clearance of chylomicrons from the blood after dietary fat ; in treated type 1 patients, abnormalities in the postprandial period may not be found .
This increased postprandial lipemia is especially marked in women, who generally have less postprandial lipemia than men. Chylomicron clearance requires several steps (Fig. 1).
After chylomicrons enter the bloodstream via the thoracic duct, apoCII, the activator of LpL, is transferred to these particles primarily from HDL. The particle then interacts with LpL on capillary lumenal endothelial cells of cardiac and skeletal muscle and adipose tissue. Released fatty acids are taken up by those tissues, perhaps via the fatty acid transporter, CD36 , and a smaller triglyceride-depleted particle, a chylomicron remnant, is created. Chylomicrons contain a truncated form of apoB termed apoB48. This protein is 48% of full-length apoB and lacks the portion of apoB that interacts with the LDL receptor. A correlation between postprandial lipemia and atherosclerosis has been found in a number of clinical studies . In addition, apoB48 remnants are found in a number of atherogenic animal models made with diets and genetic modifications . It is generally accepted that remnant lipoproteins, in addition to LDL, are atherogenic.
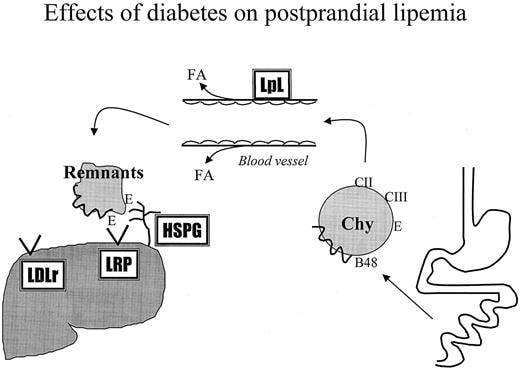
Effects of diabetes on postprandial lipemia. A defect in removal of lipids from the bloodstream after a meal is common in patients with diabetes. Chylomicron metabolism requires that these lipoproteins obtain apoCII after they enter the bloodstream from the thoracic duct. Triglyceride within the particles can then be hydrolyzed by LpL, which is found on the wall of capillaries. LpL activity is regulated by insulin, and its actions are decreased in diabetes. Triglyceride-depleted remnant lipoproteins are primarily degraded in the liver. This requires them to be trapped by liver heparan sulfate proteoglycans (HSPG) and then internalized by lipoprotein receptors, LDL receptor and LRP.
Because remnants contain a truncated form of apoB, apoB48, that does not interact with these receptors, this uptake is mediated by apoE.
Remnant lipoproteins can be removed from the bloodstream via several pathways, some of which appear to be modulated by diabetes. Liver is the major, although not exclusive, site of remnant clearance. As these particles percolate through the liver, they are trapped by association with the negatively charged proteoglycans within the space of Disse. This process may be aided by the presence of apoE and hepatic lipase, proteins that bind to both lipid particles and proteoglycans. Both hepatic lipase and heparan sulfate proteoglycan production may be reduced in diabetes. The second step in remnant clearance is via cellular internalization and degradation of the particles. Some of the remnants may be directly internalized along with cell surface proteoglycans. Most remnant uptake is via receptors. ApoE is a ligand for both the LDL receptor and LRP. Lipase enzymes (LpL and hepatic lipase) also interact with the LRP. In very poorly controlled diabetes LDL receptors may be decreased. Although LRP may be regulated by insulin in cultured macrophages (30), liver LRP is not decreased in diabetic mice .
Although most patients with poorly controlled diabetes develop hypertriglyceridemia, occasional patients develop severe hyperchylomicronemia. Triglyceride levels exceeding 1000 mg/dL lead to visibly lipemic serum. At higher levels the patients can develop eruptive xanthomas, lipemia retinalis, and pancreatitis. Most of these patients have an underlying lipid disorder, such as heterozygous LpL deficiency, that is then exacerbated by diabetes .
The relationship between severe hypertriglyceridemia and diabetes is sometimes obscured because primary LpL deficiency can lead to recurrent pancreatitis and insulin deficiency. In contrast to this, recent experimental data have shown that the LpL is expressed in the islet cells, and it has been postulated that this enzyme may promote fat-induced toxicity leading to defective insulin secretion .
Increased plasma VLDL. Patients with diabetes, especially type 2 diabetes, have increased VLDL production . Insulin infusion will correct this abnormality either because of the concomitant reduction in plasma fatty acids or because of direct effects of insulin on the liver (Fig. 2).
Effects of diabetes on VLDL production. Poorly controlled type 1 diabetes and type 2 diabetes are associated with increased plasma levels of VLDL. Two factors may increase VLDL production in the liver: the return of more fatty acids due to increased actions of hormone-sensitive lipase (HSL) in adipose tissue and insulin actions directly on apoB synthesis. Both of these processes will prevent the degradation of newly synthesized apoB and lead to increased lipoprotein production. VLDL, like chylomicrons, requires LpL to begin its plasma catabolism, leading to the production of LDL or the return of partially degraded lipoprotein to the liver.
Both the composition and the size of VLDL determine its metabolic fate. In diabetes greater amounts of fatty acids returning to the liver are reassembled into triglycerides and secreted in VLDL.
A greater content of triglyceride leads to the production of larger particles. Not all VLDL are equally likely to be converted to LDL. A greater proportion of large lighter VLDL return to the liver without complete conversion to LDL ; this pathway is akin to that of chylomicrons. Like chylomicrons, apoE may be the ligand that mediates liver uptake of these particles. Thus, VLDL metabolism is a competition between liver uptake of partially catabolized lipoproteins and intracapillary lipolysis, a process that may require several steps to complete VLDL conversion to LDL.
LDL are not usually increased in diabetes. In part this may represent a balance of factors that affect LDL production and catabolism. A necessary step in LDL production is hydrolysis of its precursor VLDL by LpL. A reduction in this step due to LpL deficiency or excess surface apoproteins (C1, C3, or possibly E) decreases LDL synthesis. Conversely, increases in this lipolytic step that accompany weight loss, fibric acid drug therapy, and treatment of diabetes may increase LDL levels. In diabetes a reduction in LDL production may be counterbalanced by decreases in LDL receptors and/or the affinity of LDL for those receptors. Both glycosylated LDL and small, dense LDL bind to LDL receptors less avidly than does normal LDL. Occasionally diabetic patients, especially those with very poor glycemic control, may have increased LDL that is reduced by treatment of their diabetes. This is due to effects on either the LDL or the receptor.
Increased small dense LDL. Heterogeneity exists in the size and composition of all classes of lipoproteins. The ratio of lipid to denser protein varies, and this determines both the buoyancy and the size of the particle, as the lipids are primarily contained in the core. In the case of VLDL and HDL, the particles also differ in their content of apoproteins, especially in the amounts of apoCs and apoE on the particle. The core of all lipoproteins contains hydrophobic cholesteryl ester and triglyceride. The proportions of these lipids are determined by CETP-mediated exchange of lipids (Fig. 3) and the actions of lipases that remove triglyceride by converting it into monoglycerides, glycerol, and free fatty acids. In the absence of a defect in these enzymes, lipoproteins enriched in triglyceride will be converted to small, denser forms. This is true for both HDL and LDL.
Plasma lipid exchange. In the presence of increased concentrations of VLDL in the circulation, CETP will exchange VLDL triglyceride for cholesteryl ester in the core of LDL and HDL. This triglyceride can then be converted to free fatty acids by the actions of plasma lipases, primarily hepatic lipase. The net effect is a decrease in size and an increase in density of both LDL and HDL.
A decrease in the size and an increase in density of LDL are characteristic of most hypertriglyceridemic states, including diabetes. Because of this, small dense LDL is considered by many to be one of the hallmarks of diabetic dyslipidemia rather than the expected companion of reduced HDL and increased triglyceride levels . The special designation given to LDL size, rather than HDL and VLDL size, is based on a large amount of clinical and experimental data implying that these particles confer additional atherosclerotic risk. In vitro, small dense LDL can be oxidized more easily, the particles do not interact with LDL receptors as well, and they may associate with proteoglycans on the surface of cells or in matrix more readily.
Although several human studies imply that small dense LDL are an additional marker for atherosclerosis development, this observation may be restricted to patients with increased levels of apoB and decreased HDL. In other studies the concomitant association of hypertriglyceridemia and low HDL appears to obscure any additional risk profiling attributable to LDL size. In dietary studies using primates, larger, not smaller, LDL size correlates with atherosclerosis, presumably because each of these LDL carries more cholesterol.
Although one could question the need to search for additional risk factors in diabetic patients who are clearly at increased risk of disease, many clinicians and research centers do measure LDL density and/or size. This can be done by measuring LDL density using an ultracentrifuge or by measuring size using gradient gels or light scattering. Another method of determining the likelihood of a patient having small dense LDL is by waist measurement, a cheaper and easier test . Obesity and insulin resistance are highly correlated with small dense LDL.
Reduced HDL. There are several reasons for the decrease in HDL found in patients with diabetes (Fig. 4).
Increased concentrations of plasma VLDL drive the exchange of triglyceride from VLDL for the cholesteryl esters found in HDL. Thus, the etiology of the hypertriglyceridemia and reduced HDL can be accounted for; CETP-mediated exchange of VLDL triglyceride for HDL cholesteryl esters is accelerated in the presence of hypertriglyceridemia. Clinical measurements of HDL are of HDL cholesterol; therefore, substitution of triglyceride for cholesteryl ester in the core of the particle leads to a decrease in this measurement. Moreover, the triglyceride, but not cholesteryl ester, in HDL is a substrate for plasma lipases, especially hepatic lipase that converts HDL to a smaller particle that is more rapidly cleared from the plasma . Another contributor to HDL is the surface lipid from triglyceride-rich particles that are transferred to HDL during VLDL and chylomicron lipolysis. This increases HDL lipid content. Defective lipolysis leads to reduced HDL production.
Effects of diabetes on HDL metabolism. HDL production requires the addition of lipid to small nascent particles. This lipid arrives via hydrolysis of VLDL and chylomicrons with transfer of surface lipids [phospholipid (PL) and free cholesterol (FC)] via the actions of phospholipid transfer protein (PLTP). A second pathway is via efflux of cellular free cholesterol (FC), a process that involves the newly described ABC1 transporter and esterification of this cholesterol by the enzyme lecithin cholesterol acyl transferase (LCAT).
HDL catabolism may occur through several steps. Hepatic lipase and scavenger receptor-BI are found in the liver and in steroid-producing cells. HDL lipid can be obtained by these tissues without degradation of entire HDL molecules. In contrast, the kidney degrades HDL protein (apoAI) without lipid, perhaps by filtering nonlipid-containing protein.
Within the last 2 yr a number of additional enzymes and receptors have been discovered that are integral regulators of HDL metabolism and presumably the effects of HDL on atherosclerosis. It is not yet clear whether hyperglycemia or insulin is an important regulator of these molecules. One of the first steps in HDL production is the addition of lipid to the small, newly formed HDL particles manufactured in the liver and intestine. Phospholipid transfer protein may be required for lipid transfer from triglyceride-rich lipoproteins.
In addition, newly formed HDL receive cholesterol from nonhepatic tissues. Theoretically, the most important of these tissues for atherosclerosis development should be the arterial wall and lipid-rich vessel macrophages. Several groups have recently identified the gene responsible for Tangier disease, a rare defect associated with very low levels of HDL and deposits of cholesterol in the tonsils and other lymphoid tissues. ABC1, a member of a family of ATP-binding cassette transporters, is defective in this disease . This protein appears to be necessary for transfer of excess cholesterol out of cells and into HDL. Cholesterol is an amphipathic molecule that would be expected to remain on the surface of a lipoprotein. Lecithin acyl transferase converts cholesterol into its hydrophobic ester form, allowing it to enter the core of the lipoprotein particle.
Unlike LDL, but more akin to triglyceride-rich lipoproteins, HDL protein and lipid metabolism are sometimes disparate. Cholesterol is the substrate for steroid hormones and bile. Liver, adrenal, and gonads can obtain HDL lipid without uptake and degradation of the entire lipoprotein. This process involves scavenger receptor-BI. By controlling the return of cholesterol to the liver, this receptor appears to play an antiatherogenic role in models of mouse atherosclerosis . Kidneys are a major site of degradation of apoAI, the major protein component of HDL. This appears to occur due to filtration of this 22-kDa protein when it is freed from HDL lipid. Fatty acids may be important for this effect; these fatty acids may be derived from hepatic lipase hydrolysis of HDL triglyceride.
Relationship of diabetic dyslipidemia to atherosclerotic risk. Trials of glucose reduction have confirmed that glucose control is the key to preventing microvascular diabetic complications. These trials have, however, failed to show a marked benefit of glucose control on macrovascular disease. There are several reasons why this could have occurred. The time course of the effects of diabetes on diseases of large arteries and small vessels differs , and longer trials may be needed. Reversal of underlying vascular disease may require a different degree of control or may follow a different time course than that for small vessels. Finally, the pathological processes are probably different. Small vessel disease of diabetic patients occurs in both type 1 and type 2 diabetes and does not occur in nondiabetics. It is clearly related to the defective glucose control.
A variety of animal models have been used to try to reproduce the relationship between diabetes and macrovascular disease. In a classic experiment, Duff et al. used alloxan to produce diabetes in cholesterol-fed rabbits. In a seemingly paradoxical result the diabetic rabbits had less, not more, atherosclerosis. This atherosclerosis was increased with insulin treatment. The reasons for this result are now apparent. These rabbits developed hyperlipidemia that was due in part to a marked defect in LpL. Large chylomicrons were not converted to more atherogenic remnant lipoproteins and were unable to penetrate the vessel and lead to lipid deposition . This pathophysiological situation is not reproduced in human diabetes, except for the rare situation in which patients are also LpL deficient.
Other animal studies have more closely imitated the situation in man. Limited studies have been performed in monkeys made diabetic using streptozotocin; in some studies the monkeys have increased LDL retention and reduced HDL.
Alloxan-treated pigs develop diabetes and increased atherosclerosis ; however, plasma LDL was more than doubled by the diabetes. Thus, the effects of diabetes cannot be discerned, because increased lipoprotein levels alone should increase atherosclerosis.
Within the past decade, genetic manipulation has made mice the most widely used animal for the study of human disease. For this reason, several investigative groups have studied the effects of hyperglycemia on atherosclerosis progression. Except for a small increase in lesions in BALB/c mice, most nontransgenic strains of mice do not have diabetes-induced atherogenesis ; most importantly, atherosclerosis was not increased in C57BL6 mice fed an atherogenic diet. There are three well defined mouse models of atherosclerosis, and all have been studied under diabetic conditions. Park et al. found that diabetes increased lesion size in diabetic mice deficient in apoE0, an effect that was inhibited by the infusion of soluble fragments of the receptor for advanced glycosylation end products. In these mice the diabetes markedly increased circulating cholesterol levels, perhaps due to a decrease in liver uptake of remnant lipoproteins via the proteoglycan-mediated pathway . Therefore, the secondary hyperlipidemia, rather than effects of the diabetes itself, might have been the primary reason for the increased atherosclerosis.
Diabetic LDL receptor knockout mice do not have more atherosclerosis than control mice. Mice that contain a transgene for expression of human apoB are more hyperlipidemic than wild-type animals and develop atherosclerotic lesions when fed a diet similar to that eaten by inhabitants of northern Europe and North America. Addition of diabetes using streptozotocin and by crossing with brown adipose tissue-deficient mice did not increase atherosclerosis in these mice . If one were convinced that hyperglycemia alone is responsible for accelerated atherosclerosis, it would appear that the mouse, despite its production of AGEs, is resistant to diabetic macrovascular disease. An alternative hypothesis that is compatible with the known human data and is consistent with the mouse and other animal models is that diabetes-mediated acceleration of vascular disease requires some additional factors missing in the mouse model. One such factor is diabetic dyslipidemia.
Treatment of dyslipidemia in patients with diabetes. There are two reasons to specifically correct lipoprotein abnormalities in patients with diabetes. These are to prevent pancreatitis due to severe hypertriglyceridemia and to reduce the risk of macrovascular complications. A number of recent reviews have focused on the use of lipid-lowering medications in diabetic patients . The objectives of that therapy will be discussed here.
The American Diabetes Association has published clinical goals for lipoprotein levels in adults with diabetes . They are as follows: optimal LDL cholesterol levels less than 100 mg/dL (2.60 mmol/L), optimal HDL cholesterol levels more than 45 mg/dL (1.15 mmol/L), and desirable triglyceride levels less than 200 mg/dL (2.3 mmol/L). The rationale for the LDL recommendation is based on the observations that adult patients with diabetes and no overt macrovascular disease appear to have the same risk of development of cardiac events as nondiabetics who already have had a cardiac event . The current National Cholesterol Education Program goal for patients with coronary heart disease is LDL levels below 100 mg/dL. Most importantly, there are available medications that should allow practitioners to reach this goal in most patients. Moreover, data exist showing that statin drugs are efficacious for LDL-lowering and disease prevention in diabetic patients.
The second goal is to increase HDL to 45 or greater. Although this may be an ideal goal, for many patients and their physicians it is not a practical one. This is acknowledged in the American Diabetes Association report . Unlike for LDL, there are limited options to achieve this goal, especially in patients with diabetic dyslipidemia who begin with HDL cholesterol levels below 35 mg/dL. Exercise, weight loss, and smoking cessation all increase HDL. Diets low in cholesterol and saturated fat tend to decrease HDL. The most effective single medication to raise HDL is niacin . A good response to this medication is an increase in HDL of 25%, which is still not enough to raise many low HDL levels to the goal. Although niacin can be given to diabetic patients, it is generally avoided because it causes worsening hyperglycemia. Fibric acids and statins also increase HDL; however, their effects are more modest that those found with niacin. Two recent intervention trials showed effective methods to reduce cardiac disease in subjects with low HDL. Neither method raised HDL to the ADA goal, nor did the studies use medications that are likely to achieve this goal in most patients. In one study subjects with HDL below 50 were treated with statins; lower LDL was associated with fewer cardiac events .
In the second, termed VA-HIT , patients with cardiac disease and average HDL of 31 mg/dL were treated with gemfibrozil, leading to a 7% HDL increase, approximately 25% triglyceride reduction, and fewer recurrent events. Therefore, it is this author’s opinion that to set a goal for HDL at 45 mg/dL is impractical, and the benefits of such a goal are unproven.
Triglyceride levels below 200 mg/dL are termed desirable; this appears to differentiate this from a goal. The primary and in many cases essential approach to triglyceride reduction is glycemic control. In type 2 patients this also means weight reduction. Although severe hypertriglyceridemia leads to increased risk for pancreatitis, proof that reduction of triglycerides is of benefit is lacking. Several investigators quote the VA-HIT trial and several subgroup analyses of fibric acid studies as evidence that treatment of elevated triglycerides is beneficial. Triglycerides can be reduced with niacin, fibric acids, high dose statins, and fish oil. It should be noted that the use of fibric acids to reduce triglyceride along with statins increases the risk of myositis and should be used with caution