Renal cell carcinoma (RCC) is the sixth most common solid organ cancer in the UK. In 2018, there were 403,262 people diagnosed worldwide with the disease (2.2% of all cancer cases), and it accounted for 175,098 deaths in total (1.8% of all cancer deaths) [1]. The abundant use of cross-sectional imaging in modern medical practice has led to an increased detection of small renal masses of uncertain clinical significance. These range from simple renal cysts requiring no follow-up to malignant renal carcinomas requiring intervention.
The majority of patients with renal cancer present with localised disease and the mainstay of treatment is surgical excision. Up to 30% of patients will present with metastatic disease and a further 25-40% of those treated with curative intent will go on to develop metastatic disease [2]. Encouragingly there have been significant developments in the management of metastatic disease in recent years. In this review, we will focus on the changing role of immunotherapy in the management of metastatic RCC (mRCC).
“Combining checkpoint inhibitors can work in a synergistic manner, limiting doses and potentially lowering toxicity”
Cytokine immunotherapy in RCC
The role of immunotherapy in cancer treatment dates back to 1891, when William B Coley (an American bone surgeon) injected streptococcal organisms into a patient with inoperable cancer leading to its shrinkage. He continued with this practice, injecting over 1000 cancer patients with bacteria or bacterial products. Although ‘Coley’s Toxins’ were met with widespread criticism at the time, this was effectively the first use of immunotherapy in the treatment of cancer [3].
Historically, RCC was recognised as an immune regulated disease as tumours are rich in immune infiltrates. Cases have been described of spontaneous regression of RCC proven on biopsy. Although this is extremely rare, it is thought to be regulated by an immune process [4]. Early studies of utilising immune modulators focused on ‘non-specific immunotherapy’ – with the cytokines interferon-α and interleukin-2 being the first compounds demonstrating positive results.
Interferon-α
Interferon-α was the first cytokine based compound used in the treatment of mRCC. Interferons play an integral role in the immune response, albeit not a fully understood one. Interferon-α is a pleiotropic cytokine with immunomodulary, anti-viral, anti-proliferative and anti-angiogenic properties. Its anti-tumour effect is based on its ability to induce the differentiation of monocytes into highly activated dendritic cells. Highly activated dendritic cells recognise complex antigens and induce both T and B-cell immunity. T-cell immunity being important for the anti-tumour process.
Early studies demonstrated overall response rates to interferon-α ranging from 0-29%, however larger studies reported a median overall survival of approximately 15% [5]. The best results for interferon-α as monotherapy are achieved in patients with good performance status and pulmonary metastases upon surgery [6]. The main limitation with interferon-α therapy was that it was poorly tolerated, with flu-like side-effects such as fatigue, fever, myalgia and depression [7].
Interleukin-2
Interleukin-2 (IL-2) is another compound that plays a central role in the immune system. IL-2 is a cytokine that is secreted by activated T-cells (both CD4+ and CD8+), natural killer cells and dendritic cells. It is recognised as a T-cell growth factor, and can increase T-cell proliferation and differentiation. The IL-2 receptor is highly expressed on activated T-cells, and so the ligation of IL-2 with the IL-2 receptor leads to proliferation and differentiation of B and T-cells and stimulation of a mass of other cytokines. Activation of the IL-2 receptor initiates signal transduction through JAK3, STAT5, MAPK and PI3K pathways with the activation of these pathways affecting gene expression and altering cell growth and immune function. The anti-tumour effect of IL-2 is based on its ability to cause proliferation of natural killer cells (NK), lymphokine-activated killer cells and other cytotoxic cells [8].
In 1995 Fyfe et al. showed high dose IL-2 induced durable long-term remissions in approximately 10% of patients with previously untreated mRCC. A non-randomised series of 255 patients showed 5% (12) had complete responses and 9% (24) had partial responses. However, IL-2 treatment was poorly tolerated and also associated with a treatment related death rate of 4% [9].
![]()
With greater understanding of the pathogenesis of RCC and in particular the clear cell subtype, targeted therapy focused on disruption of the VHL-HIF pathway was developed. Tyrosine kinase inhibitors (TKI) became first-line treatment of RCC following the phase III trial comparing sunitinib to interferon demonstrating a six-month median progression-free survival (PFS) benefit for sunitinib with favourable side-effect profile (PFS 11 months and 5 months respectively) [10]. Subsequent development and introduction of other TKIs and mTOR inhibitors ensued, however despite the improved survival outcomes they lacked the reports of complete response demonstrated by these systemic immunotherapies.
Attempts to combine interferon-α with bevacizumab, a vascular endothelial growth factor (VEGF) antibody resulted in no improvement in overall survival compared with interferon-α alone [11]. However, appreciation of the role of the immune checkpoint in tumour evasion from the immune system and manipulation of this response for survival benefit in other tumour types has led to the study and use of immune checkpoint inhibitors in the management of mRCC.
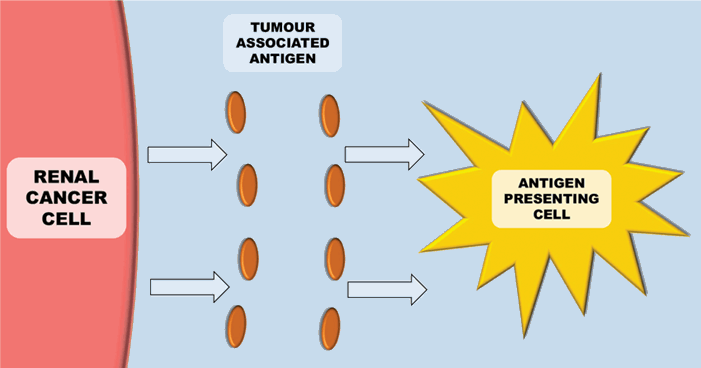
Figure 1: Antigen presenting cells (e.g. Dendritic cells) recognise tumour antigens.
Immune checkpoint pathways
Mechanism of immune checkpoints
The immune system plays a key role in controlling and eliminating cancer – especially RCC. The process by which this happens is quite complex. Antigen-presenting cells (APCs) (e.g. dendritic cells), recognise tumour associated antigens which are expressed on tumour cells (Figure 1). These then present the tumour associated antigens on their surface within a major histocompatibility complex (MHC) (either class I or class II). T-cell receptors then bind to the MHC complex on the antigen presenting cell (which carries the tumour associated antigen). In order for the T-cell to become activated, it requires co-stimulation by binding CD28 receptor on T-cells to either CD80 or CD86 (Figure 2). This ligation then leads to a cascade of processes leading to the cytotoxic effect of CD8+ T-cells on cancer cells.
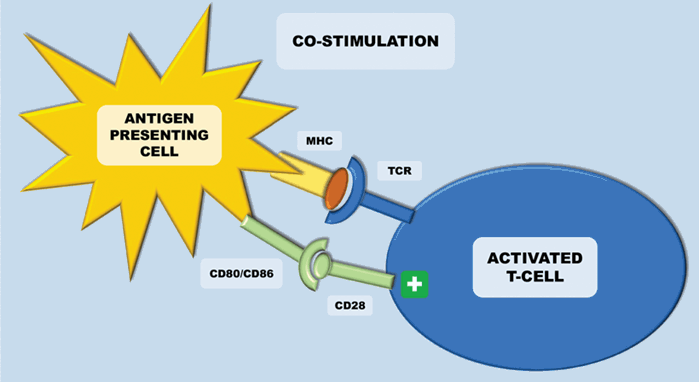
Figure 2: Antigen presenting cells (APCs) present the tumour associated antigens on their surface within a major histocompatibility complex (MHC) – this is either class I or class II. In order for the T-cell to become activated, it requires co-stimulation by binding CD28 receptor to either CD80 or CD86 (both B7 ligands), which are released upon maturation of APCs. This leads to a cascade of processes eventually leading to the cytotoxic effect of CD8+ T-cells on cancer cells.
In the setting of malignancy, multiple mechanisms of immune suppression may exist that prevent effective anti-tumour immunity. Immune ‘checkpoints’ consist of multiple co-stimulatory and inhibitory interactions, which sustain self-tolerance and modulate physiological immune responses. The lack of co-stimulatory signals on the antigen presenting cell blocks the cytotoxic activation of T cells by the antigen. This is termed a tolerogenic signal. Amongst others, two important regulators of this are cytotoxic-T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1). They are expressed on T-cells, regulatory T-cells and myeloid derived suppressor cells (MDSC) and they inhibit T-cells in order to regulate excessive immune response. Immune checkpoint pathways – such as PD-1 and CTLA-4 - are co-opted by cancer, resulting in altered expression of proteins to assist in the masking of cancer cells from immune surveillance and thus to evade immune destruction [12]. Modulation of these checkpoints can lead to the targeting and destruction of cancer cells with the preservation of normal cells.
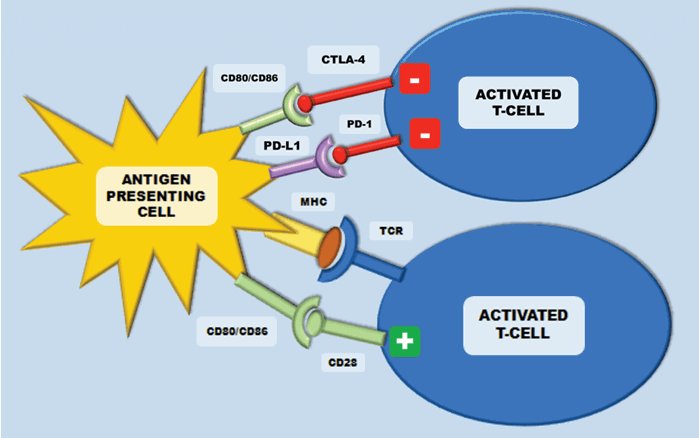
Figure 3: The CTLA-4 receptor is inhibitory and is expressed following T-cell activation. This terminates CD-28 mediated T-cell activation. In cancer, CTLA-4/CD28 engagement down-modulates helper T-cell activity and enhances regulatory T-cells immunosuppressive activity. Programmed cell death protein-1 (PD-1) is also expressed on activated T-cells and inhibits immune response upon response. This inhibition is initiated after binding to the ligands PD-L1 and PD-L2.
CD28/CTLA-4 system
CTLA-4 is an inhibitory receptor, which is expressed exclusively on T-cells. In humans, polymorphisms of the CTLA-4 gene have been associated with auto-immune conditions such as type-1 diabetes. It is expressed on both CD4+ helper T-cells and CD8+ cytotoxic T cells, however it is predominantly expressed on CD4+ helper cells. During early T cell activation, it is expressed and transferred to the plasma membrane. CTLA-4 binds the CD28 receptors with a much higher affinity than the B7 ligands from the antigen presenting cell – this terminates the CD-28 mediated T-cell activation and subsequent IL-2 production (Figure 3). In cancer, CTLA-4/CD28 engagement down-modulates helper T-cell activity and enhances regulatory T cells immunosuppressive activity. If CTLA-4 is inhibited, there are two main results – inhibition of peripheral T-cell tolerance resulting in autoimmunity and activation of anti-tumour immunity [12] (Figure 4).
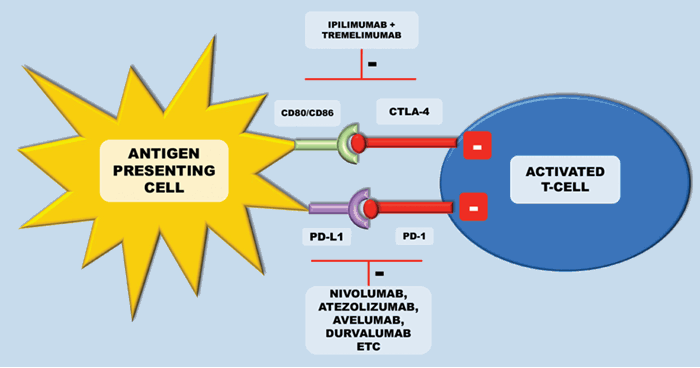
Figure 4: Inhibition of these checkpoints can lead to activation of anti-tumour immunity.
Currently, licensed anti-CTLA-4 therapy includes ipilimumab (a fully human IgG1 anti-CTLA-4 monoclonal antibody) and tremelimumab (a fully human IgG2 anti-CTLA-4 monoclonal antibody). In metastatic melanoma – ipilimumab has become a standard of care, improving overall survival in those with previously treated metastatic melanoma [13]. In mRCC the results with ipilimumab were less satisfactory. A phase II study evaluated the response of ipilimumab, with two cohorts receiving either 3mg/kg followed by 1mg/kg or all doses at 3mg/kg every three weeks. Only one patient of the 21 patients receiving the lower dose had a partial response, and five of the 40 patients at the higher dose had a partial response. A third of patients experienced either a grade III or grade IV immune mediated toxicity. Notably there was a significant association between auto-immune toxicity and tumour regression [14]. Toxicty related to anti-CTLA-4 therapy is secondary to systemic inflammation leading to problems such as colitis, hepatitis and occasionally severe pneumonitis.
PDL1 / PD1 system
Programmed cell death protein-1 (PD-1) is also a transmembrane receptor that is expressed on activated T-cells to inhibit immune response upon activation by antigens presented on the APCs. This inhibition is initiated after binding to the ligands PD-L1 and PD-L2 (Figure 3). PD-L1 is expressed in tumour cells and tumour-infiltrating lymphocytes to stop activated CD4+ and CD8+ T-cells targeting the tumour cells. Inhibiting this mechanism should then lead to tumour regression (Figure 4). In RCC, PD-L1 expression on either tumour cells or tumour-infiltrating lymphocytes in primary tumours correlates with a worse prognosis [15]. Targeted therapies against PD-1 receptor and its ligand PD-L1 have demonstrated impressive response rates with minimal toxicity in several malignancies. Melanoma, ovarian and lung cancer biopsies have all been shown to have high PD-L1 expression levels.
Nivolumab was the first antibody directed against PD-1 showing anti-tumour activity in RCC. CheckMate-025 was a phase III study that randomised 821 previously treated patients to receive nivolumab or everolimus (i.e. second or third line treatment). Median overall survival was 25 months for nivolumab versus 19.6 months for everolimus. Interestingly, benefits were seen regardless of PD-L1 expression. In addition to this, side-effect profile favoured treatment with nivolumab and more patients had clinically meaningful improvement in health-related Quality of Life (QOL) scores. A number of other licensed drugs target PD-1. Both nivolumab and pemrolizumab are licensed to treat a number of tumour types including non-small cell lung cancer, melanoma, head and neck cancer, urothelial bladder cancer, RCC and Hodgkins lymphoma [8].
In addition to targeting PD-1, antibodies targeting the PD-L1 receptor are in development or licensed for other indications. Atezolizumab is licensed in the treatment of advanced urothelial cancer and phase 1a study has shown a manageable safety profile and promising anti-tumour activity in patients with mRCC [16]. Avelumab and durvalumab are in late stage clinical development for a number of indications [17]. The JAVELIN Solid Tumour trial, a phase I trial of avelumab has shown acceptable safety of the drug in patients with advanced solid tumours and clinical activity in patients with gastric or gastroesophageal junction cancer and disease progression after chemotherapy [18]. In 2014, a phase II study of avelumab (JAVELIN Merkel 200) enrolled patients with Merkel cell carcinoma progressed after chemotherapy.
Recently published data from this study showed an objective response rate of 31.8%, with 9.1% achieving complete remission, and responses ongoing in 82% of responders. This demonstrated the efficacy of avelumab in this aggressive disease and the US Food & Drug Administration (FDA) have since approved avelumab for this indication [19]. A phase III trial is currently ongoing assessing avelumab in combination with axitinib for mRCC (JAVELIN Renal 101; NCT02684006). In addition to this, tremelimumab combined with durvalumab (a PD-L1 inhibitor) have been shown to have a manageable safety profile in a variety of advanced solid tumours, including mRCC, in a phase 1 trial [14].
Combination therapy
Combining checkpoint inhibitors can work in a synergistic manner, limiting doses and potentially lowering toxicity. In RCC, atezolizumab and durvalumab have demonstrated promising results in phase I trials – and are undergoing tests in combination with VEGF or other checkpoint inhibitors. The IMmotion 151 study is a phase III combination trial. In this, a total of 915 patients were randomised to treatment with atezolizumab plus bevacizumab versus sunitinib (a tyrosine kinase inhibitor). Endpoints were progression-free survival in PD-L1+ patients (defined as >1% on tumour infiltrating immune cells), and overall survival in the intention to treat population. Median progression-free survival in the PD-L1+ group was 11.2 months in the combination group versus 7.7 months in the sunitinib arm – with similar results seen for progression-free survival in the intention to treat analysis. However, an assessment by the independent radiology committee deemed no significant improvement was seen. Overall survival data were immature at first readout. Of note, the combination of atezolizumab and bevacizumab was well tolerated with a favourable toxicity profile compared with sunitinib, and only 15% of patients of patients treated with combination required corticosteroids within 30 days of an immune mediated adverse reaction [20].
CheckMate-124 evaluated the combination of ipilimumab and nivolumab in combination versus sunitinib – endpoints being overall survival, progression-free survival and objective response rate amongst patients with an intermediate or poor-risk prognosis by International Metastatic RCC Database Consortium (IMDC) criteria. Median OS was not reached in the combination arm versus 26 months with sunitinib. There was a numerical improvement in PFS in the combination group, but this was not statistically significant. The improved overall survival with the combination was seen regardless of PD-L1 expression, although the extent was more pronounced in this group. Combination therapy had a complete response rate of 9% compared with 1% in sunitinib – in the context of an improved objective response rate with the combination.
Analysis of favourable risk patients, found that progression-free survival and objective response rate favoured sunitinib in this subgroup. Toxicity was greater in the combination group, with adverse effects leading to discontinuation occurring in 22% in the combination group and 12% in the sunitinib group [21]. In April 2018, the US FDA approved the combination of ipilimumab and nivolumab in untreated patients with intermediate or poor risk mRCC. The European Medicines Agency (EMA) approved the combination in November 2018 and subsequently the European Association of Urology (EAU) guidelines recommended this treatment in the same group of patients.
There are multiple ongoing clinical trials investigating combinations of checkpoint inhibitors and anti-VEGF therapies. Nivolumab, atezolizumab, avelumab and pembrolizumab are all currently undergoing investigation combined with various anti-VEGF agents, ranging from pilot studies to phase III trials, and in first and second-line settings.
Adjuvant therapy
Studies investigating the effective of adjuvant TKI therapy or mTOR inhibitors in patients with localised RCC after resection with high risk of recurrence have failed to demonstrate oncological benefit. Given the difference in mechanism, there is renewed hope that checkpoint inhibitors may offer benefit to this group of patients. To this end, there are four currently recruiting randomised clinical trials investigating the oncological effect of atezolizumab (NCT03024996), pembrolizumab (NCT03142334), durvalumab or combination durvalumab and tremelimumab (NCT03288532) or combination nivolumab and ipilimumab (NCT03138512) following surgical excision of high or intermediate-risk localised RCC. It is likely to be a number of years before these trials complete recruitment and report progression-free or cancer-specific survival.
Alternative immunotherapies
Although CTLA-4 and PD-1 inhibitors are currently the most promising uses of immunotherapy in mRCC, there are other possible targets for ‘checkpoint’ inhibition with research ongoing in tumour immunology. Current areas of research include antibodies to inhibit or activate T-cells to treat solid tumours. In RCC, MHC class II agonists have shown promise in phase I trials, with the absence of toxicity and demonstration of activity. They work by activating dendritic cells and subsequently T-cells [22]. Other avenues are antibodies against co-stimulatory tumour necrosis family receptors (TNFR) [23].
Conclusion
The management of renal cancer is complex and is a rapidly evolving arena, with the role of immunotherapy depending on the stage of disease. There is clear evidence of benefit with selected checkpoint inhibitors in the treatment of metastatic RCC. This is reflected in current EAU guidelines, which recommend the use of ipilimumab plus nivolumab in treatment naïve patients with mRCC who are intermediate and poor risk. Bevacizumab and interferon-α could be considered as a second line treatment for patients with advanced RCC. Nivolumab is also recommended as second-line treatment in patients who have previously been treated with VEGF-targeted therapy. With ongoing trials investigating combination treatments, this is likely to change further over coming years. There is no evidence for the use of checkpoint inhibitors in the adjuvant setting although we await the outcomes of currently recruit trials with excitement.