The right therapy for lupus retinal vasculitis can be sight- or life-saving, according to Paula Pecen, MD, a vitreoretinal surgery fellow at Cleveland Clinic’s Cole Eye Institute.
Lupus retinal vasculitis may lead to significant vision loss if left untreated, and can be an indicator of underlying systemic disease, including lupus cerebral vasculitis. This is rare, but can lead to cerebral infarction. It is crucial to differentiate lupus retinal vasculitis from standard vascular occlusion syndromes.
Case study: Patient presentation
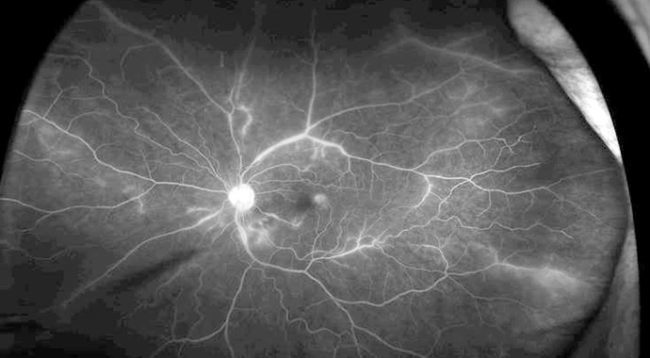
A 52-year-old Caucasian female presented with painless, blurry vision with flashes in both eyes over the past few weeks. Her past medical history included chronic kidney disease, hypertension and type 2 diabetes. She was also recently diagnosed with systemic lupus erythematosus (SLE).
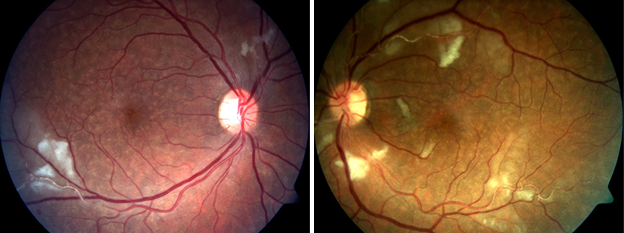
On examination, her vision was 20/20 in the right eye and 20/25 in the left eye. She had mild vitritis in both eyes, and on funduscopic examination, bilateral branch retinal artery occlusions (BRAOs) with sclerotic arteries and cotton wool spots.
Differential diagnoses
Differential diagnoses of BRAO include hypertensive retinopathy, systemic emboli, sickle cell retinopathy, Susac’s syndrome or retinal vasculitis. The many causes of retinal arteritis/vasculitis include several infectious and inflammatory ones. The former include toxoplasmosis, tuberculosis, syphilis and herpetic viruses; the latter, SLE, giant cell arteritis, granulomatosis with polyangiitis (formerly Wegener’s granulomatosis), polyarteritis nodosa, inflammatory bowel disease, sarcoidosis and multiple sclerosis.
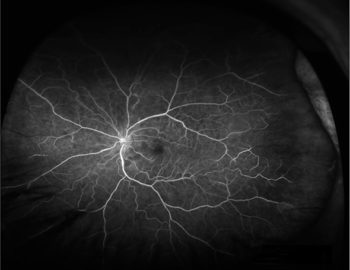
Patient diagnosis
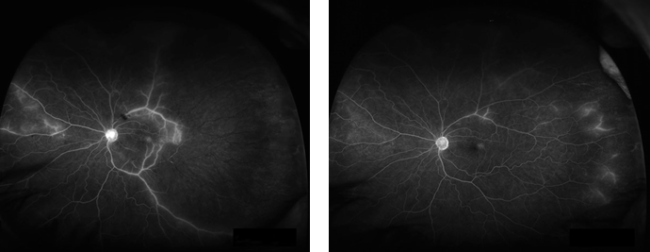
A fluorescein angiogram showed evidence of BRAOs with delayed filling and multiple areas of arteriolar leakage. An embolic workup would have been performed. However, the patient suffered a recent transient ischemic attack, and an echocardiogram showed no significant valvular calcification. A carotid ultrasound found no significant stenosis either.
Treatment
The mainstay treatment of retinal disease in lupus is systemic immunosuppression with high dose oral corticosteroids. This may be preceded by intravenous (IV) methylprednisolone if ocular findings are especially severe. Corticosteroids are then supplemented with other steroid-sparing immunosuppressive agents. It is helpful to work closely with the patient’s rheumatologist to monitor systemic side effects of medications and to monitor control of systemic lupus symptoms, according to Dr. Pecen.
The patient was initially started on immunosuppressive therapy with hydroxychloroquine (Plaquenil®) and prednisone. Hydroxychloroquine was discontinued and replaced by adalimumab (Humira®) and mycophenolate mofetil (CellCept®). The retinal vasculitis improved over a few months; this was followed by a tapering off of oral prednisone.
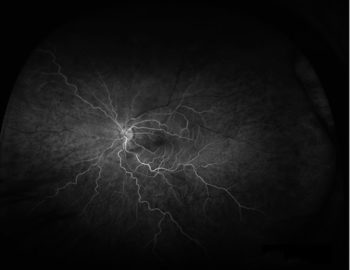
Six months later, the patient self-discontinued her immunosuppressive therapies after a series of ailments, including shingles, within a few weeks. She had a severe relapse of retinal vasculitis as well as serious joint pain in her hands that made it difficult for her to work as a baker. 590x-Inset-3b-Vascular-LeakageHigh dose IV corticosteroids were re-initiated, with transition to oral prednisone. This was followed by infliximab (Remicade®), with a suppressive dose of valacyclovir (Valtrex®). Currently, she has improved peripheral retinal perfusion and is being tapered off prednisone with good disease control on infliximab.
Follow-up
Close follow-up is needed to ensure that treatment is effective. Depending on the disease severity and manifestations, most patients with SLE should continue long-term quarterly monitoring with their rheumatologist. If a patient with SLE is initiated on immunosuppressive therapy, the treatment should not be discontinued without the oversight of a rheumatologist.