SIADH- Syndrome of Inappropriate (Increased) ADH
Hyponatremia
CLINICAL CHARACTERISTICS
Excessive secretion or action of AVP results in the production of decreased volumes of more highly concentrated urine. If not accompanied by a commensurate reduction in fluid intake or an increase in insensible loss, the reduction in urine output results in excess water retention with expansion and dilution of all body fluids. In some patients, excessive intake results from inappropriate thirst. If the hyponatremia develops gradually or has been present for more than a few days, it may be largely asymptomatic. However, if it develops acutely, it usually is accompanied by symptoms and signs of water intoxication that may include mild headache, confusion, anorexia, nausea, vomiting, coma, and convulsions. Severe hyponatremia may be lethal. Other clinical signs and symptoms vary greatly, depending on the pathogenesis of the defect in antidiuretic function.
ETIOLOGY
Hyponatremia and impaired urinary dilution can be caused by either a primary or a secondary defect in the regulation of AVP secretion or action. The primary forms are generally referred to as the syndrome of inappropriate antidiuresis .
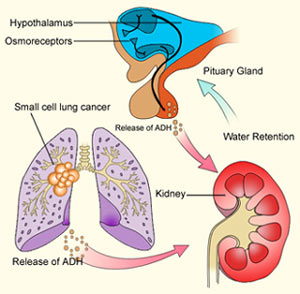
They have many different causes, including ectopic production of AVP by lung cancer or other neoplasms; eutopic release by various diseases or drugs; and exogenous administration of AVP, desmopressin, or large doses of oxytocin . The ectopic forms result from abnormal expression of the AVP-NPII gene by primary or metastatic malignancies. The eutopic forms occur most often in patients with acute infections or strokes but have also been associated with many other neurologic diseases and injuries. In this case, the SIAD is usually self-limited and remits spontaneously within 2–3 weeks, but about 10% of cases are chronic. The mechanisms by which these diseases disrupt osmoregulation are not known. The defect in osmoregulation can take any of four distinct forms .
In one of the most common ( reset osmostat ), AVP secretion remains fully responsive to changes in plasma osmolarity/sodium but the threshold, or set point, of the osmoregulatory system is abnormally low. These patients differ from those with the other types of osmoregulatory defect in that they are able to maximally suppress plasma AVP and dilute their urine if their fluid intake is high enough to reduce their plasma osmolarity/sodium to the new set point.
Another, smaller subgroup (10% of the total) has inappropriate antidiuresis without a demonstrable defect in the osmoregulation of plasma AVP . In some of them, all young boys, the inappropriate antidiuresis has been traced to a constitutively activating mutation of the V2 receptor gene. This unusual variant may be referred to as f amilial nephrogenic SIAD to distinguish it from other possible causes of the syndrome.
Causes of Syndrome of Inappropriate Antidiuresis (Siad)
Neoplasms
Carcinomas
Lung
Duodenum
Pancreas
Ovary
Bladder, ureter
Other neoplasms
Thymoma
Mesothelioma
Bronchial adenoma
Carcinoid
Gangliocytoma
Ewing’s sarcoma
Head trauma (closed and penetrating)
Infections
Pneumonia, bacterial or viral
Abscess, lung or brain
Cavitation (aspergillosis)
Tuberculosis, lung or brain
Meningitis, bacterial or viral
Encephalitis
AIDS
Vascular
Cerebrovascular occlusions, hemorrhage
Cavernous sinus thrombosis
Genetic
X-linked recessive
(V2 receptor gene)
Neurologic
Guillain-Barré syndrome
Multiple sclerosis
Delirium tremens
Amyotrophic lateral sclerosis
Hydrocephalus
Psychosis
Peripheral neuropathy
Congenital malformations
Agenesis corpus callosum
Cleft lip/palate
Other midline defects
Metabolic
Acute intermittent porphyria
Pulmonary
Asthma
Pneumothorax
Positive-pressure respiration
Drugs
Vasopressin or desmopressin
Chlorpropamide
Oxytocin, high dose
Vincristine
Carbamazepine
Nicotine
Phenothiazines
Cyclophosphamide
Tricyclic antidepressants
Monoamine oxidase inhibitors
Serotonin reuptake inhibitors
The secondary forms of osmotically inappropriate antidiuresis also have multiple causes. They usually are subdivided into three types, depending on the nature of the abnormal stimulus and the state of extracellular fluid volume.
Type I occurs in sodium-retaining, edema-forming states such as congestive heart failure, cirrhosis, and nephrosis. It is associated with markedly excessive retention of water and sodium that is thought to be stimulated by a large reduction in “effective” blood volume caused by low cardiac output and/or redistribution of plasma from the intravascular space to the interstitial space.
Type II occurs in sodium-depleted states such as severe gastroenteritis, diuretic abuse, and mineralocorticoid deficiency. It is due to stimulation of AVP by a large reduction in blood volume and/or pressure. In both types, the increased AVP secretion appears to be due to downward resetting of the osmostat.
Type III is due to nonosmotic, nonhemodynamic AVP stimuli such as nausea or isolated glucocorticoid deficiency that produce a form of euvolemic hyponatremia similar to SIAD. They are differentiated because the cause of excess AVP secretion in type III can be corrected quickly and completely by treatments (antiemetics or glucocorticoids) that are not useful in SIAD.
Differential Diagnosis of Hyponatremia Based on Clinical Assessment of Extracellular Fluid Volume (Ecfv)
Clinical Findings Type I, Hypervolemic Type II, Hypovolemic Type III, Euvolemic SIAD Euvolemic
History
CHF, cirrhosis, or nephrosis Yes No No No
Salt and water loss No Yes No No
ACTH–cortisol deficiency and/or nausea and vomiting No No Yes No
Physical examination
Generalized edema, ascites Yes No No No
Postural hypotension Maybe Maybe Maybe a No
Laboratory
BUN, creatinine High-normal High-normal Low-normal Low-normal
Uric acid High-normal High-normal Low-normal Low-normal
Serum potassium Low-normal Low-normal b Normal c Normal
Serum albumin Low-normal High-normal Normal Normal
Serum cortisol Normal-high Normal-high d Low e Normal
Plasma renin activity High High Low f Low
Urinary sodium (meq unit of time) g Low Low h High i High i
a Postural hypotension may occur in secondary (ACTH-dependent) adrenal insufficiency even though extracellular fluid volume and aldosterone are usually normal.
b Serum potassium may be high if hypovolemia is due to aldosterone deficiency.
c Serum potassium may be low if vomiting causes alkalosis.
d Serum cortisol is low if hypovolemia is due to primary adrenal insufficiency (Addison’s disease).
e Serum cortisol will be normal or high if the cause is nausea and vomiting rather than secondary (ACTH-dependent) adrenal insufficiency.
f Plasma renin activity may be high if the cause is secondary (ACTH) adrenal insufficiency.
g Urinary sodium should be expressed as the rate of excretion rather than the concentration. In a hyponatremic adult, an excretion rate >25 meq/d (or 25 ueq/mg of creatinine) could be considered high.
h The rate of urinary sodium excretion may be high if the hypovolemia is due to diuretic abuse, primary adrenal insufficiency, or other causes of renal sodium wasting.
i The rate of urinary sodium excretion may be low if intake is curtailed by symptoms or treatment.
Abbreviations: ACTH, adrenocorticotropic hormone; BUN, blood urea nitrogen; CHF, congestive heart failure; SIAD, syndrome of inappropriate antidiuresis.
PATHOPHYSIOLOGY

When osmotic suppression of antidiuresis is impaired for any reason, retention of water and dilution of body fluids occur only if intake exceeds the rate of obligatory and insensible and urinary losses. The excess water intake sometimes is due to an associated defect in the osmoregulation of thirst (dipsogenic) but also can be psychogenic or iatrogenic, including IV administration of hypotonic fluids.
In SIAD, the excessive retention of water expands extracellular and intracellular volume, increases glomerular filtration and atrial natriuretic hormone, suppresses plasma renin activity, and increases urinary sodium excretion. This natriuresis reduces total body sodium, and this serves to counteract the extracellular hypervolemia but aggravates the hyponatremia. The osmotically driven increase in intracellular volume results in swelling of brain cells and increases intracranial pressure; this is probably responsible for the symptoms of acute water intoxication. Within a few days, this swelling may be counteracted by inactivation or elimination of intracellular solutes, resulting in the remission of symptoms even though the hyponatremia persists. The pathophysiology of type III (euvolemic) hyponatremia is probably similar to that of SIAD.
In type I (hypervolemic) or type II (hypovolemic) hyponatremia, the antidiuretic effect of hemodynamically induced AVP release is enhanced by decreased distal delivery of glomerular filtrate that results from increased reabsorption of sodium in proximal nephrons. If the marked reduction in urine output is not associated with a commensurate reduction in water intake or an increase in insensible loss, body fluids are expanded and diluted, resulting in hyponatremia. Unlike SIAD, however, glomerular filtration is reduced and plasma renin activity and aldosterone are elevated due to either effective hypovolemia (type I) or absolute hypovolemia (type II). Thus, urinary sodium excretion is low (unless sodium reabsorption is impaired by a diuretic), and the hyponatremia is usually accompanied by hypokalemia, azotemia, and hyperuricemia. The sodium retention is an appropriate compensatory response to severe volume and sodium depletion in type II but is inappropriate and deleterious in type I since body sodium and extracellular volume are already markedly increased, as evidenced by the presence of generalized edema.
DIFFERENTIAL DIAGNOSIS

SIAD is a diagnosis of exclusion that usually can be made from the history, physical examination, and basic laboratory data. The possibility that hyponatremia is due to an osmotically driven shift of water from the intracellular space to the extracellular space can be excluded if plasma glucose is not high enough to account for the hyponatremia [ serum sodium decreases 1 meq/L for each rise in glucose of 2 mmol/L (36 mg/dL)] and/or plasma osmolarity is reduced in proportion to sodium (each decrease in serum sodium of 1 meq/L should reduce plasma osmolarity by 2 mosmol/L ). The type of hypotonic hyponatremia can then be determined by standard clinical indicators of the extracellular fluid volume . If these findings are ambiguous or contradictory, measuring the rate of urinary sodium excretion or plasma renin activity may be helpful provided that the hyponatremia is not in the recovery phase or due to a primary defect in renal conservation of sodium, diuretic abuse, or hyporeninemic hypoaldosteronism. The latter may be suspected if serum potassium is elevated instead of low as it usually is in types I and II hyponatremia. Measurements of plasma AVP are currently of no value in differentiating among the three types of hyponatremia since the abnormalities are similar. In patients who fulfill the clinical criteria for type III (euvolemic) hyponatremia, morning plasma cortisol should also be measured to exclude secondary adrenal insufficiency. If it is normal and there is no history of nausea/vomiting, the diagnosis of SIAD is confirmed and a careful search for occult lung cancer or other common causes of the syndrome should be undertaken. If an activating mutation of the V2receptor gene is suspected, plasma AVP should be measured while the hyponatremia and antidiuresis are present. If it is undetectable, DNA should be collected for analysis of the V2receptor gene.
TREATMENT: HYPONATREMIA
The management of hyponatremia differs depending not only on the type but also on the severity and duration of symptoms. In a patient with SIAD and few symptoms, the objective is to reduce body water gradually by restricting total fluid intake to less than the sum of urinary and insensible losses. Because the water derived from food (300–700 mL/d) usually approximates basal insensible losses in adults, total discretionary intake (all liquids) should be at least 500 mL less than urinary output. If achievable, this usually reduces body water and increases serum sodium by about 1–2% per day. If the symptoms or signs of water intoxication are more severe, the hyponatremia can be corrected more rapidly by supplementing the fluid restriction with IV infusion of hypertonic (3%) saline. This treatment also has the advantage of correcting the sodium deficiency that is partly responsible for the hyponatremia in SIAD and produces a solute diuresis that serves to remove some of the excess water. However, if plasma sodium is raised too rapidly or too much and the hyponatremia has been present for >24–48 hours, it also has the potential to produce central pontine myelinolysis , an acute, potentially fatal neurologic syndrome characterized by quadriparesis, ataxia, and abnormal extraocular movements. The risk of this complication can be minimized by observing several precautions: 3% saline should be infused at a rate >=0.05 mL/kg body weight per min; the effect should be monitored continuously by STAT measurements of serum sodium at least once every 2 hours; and the infusion should be stopped as soon as serum sodium increases by 12 mmol/L or to 130 mmol/L, whichever comes first. Urinary output should be monitored continuously since SIAD can remit spontaneously at any time, resulting in an acute water diuresis that greatly accelerates the rate of rise in serum sodium produced by fluid restriction and 3% saline infusion.
In chronic SIAD, the hyponatremia can be corrected by treatment with demeclocycline , 150–300 mg PO tid or gid, or fludrocortisone , 0.05–0.2 mg PO bid. The effect of the demeclocycline manifests in 7–14 days and is due to production of a reversible form of nephrogenic DI. Potential side effects include phototoxicity and azotemia. The effect of fludrocortisone also requires 1–2 weeks and is partly due to increased retention of sodium and possibly inhibition of thirst. It also increases urinary potassium excretion, which may require replacement through dietary adjustments or supplements and may induce hypertension, occasionally necessitating discontinuation of the treatment.
Nonpeptide AVP antagonists that block the antidiuretic effect of AVP have been used experimentally to treat SIAD. They produce a dose-dependent increase in urinary free-water excretion , that, if combined with a modest restriction of fluid intake, reduces body water and corrects the hyponatremia. The antagonists appear to have no adverse side effects, but, like hypertonic saline, they probably carry the risk of inducing osmotic demyelinization if the hyponatremia is corrected too rapidly. One of them, a combined V2/V1a antagonist ( Conivaptan ) , has been approved for short-term in-hospital IV treatment of SIAD and the hyponatremia of congestive heart failure. It is a substrate and inhibitor of cytochrome P450 and should not be used in conjunction with other drugs metabolized by these pathways. Other V2 receptor antagonists are currently in phase III trials.
In type I hyponatremia , fluid restriction is also appropriate and somewhat effective if it can be maintained. However, infusion of hypertonic saline is contraindicated because it further increases total body sodium and edema and may precipitate cardiovascular decompensation. Preliminary studies with antagonists of V2 receptors indicate that they are almost as effective in type I hyponatremia as they are in SIAD.
In type II hyponatremia , the defect in AVP secretion and water balance usually can be corrected easily and quickly by stopping the loss of sodium and water and/or replacing the deficits by mouth or IV infusion of normal or hypertonic saline . As with the treatment of other forms of hyponatremia, care must be taken to ensure that plasma sodium does not increase too rapidly. Fluid restriction and administration of AVP antagonists are contraindicated in type II as they would only aggravate the underlying volume depletion and could result in hemodynamic collapse.
In euvolemic hyponatremia due to protracted nausea and vomiting or isolated glucocorticoid deficiency ( type III ), all abnormalities can be corrected quickly and completely by giving an antiemetic or stress doses of hydrocortisone . As with other treatments, care must be taken to ensure that serum sodium does not rise too quickly or too far.